
After the introductory posts about organic solar cells – split in parts zero, one and two, – I would like to present a somewhat more intuitive picture today… well, picture indeed says it all;-)
Step 1: Light Absorption => Exciton Generation
 |

|
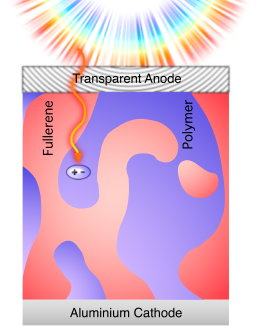
- light is absorbed in the donor material, e.g., a conjugated polymer
- excitons are thus created, strongly bound electron-hole pairs on the polymer chain
- very high absorption coefficient, device thickness on ~100nm scale, as compared to the inorganic polycrystalline semiconductor CuInSe2 (~1 micron) and crystalline Silicon (~100 micron)
- but: only narrow absorption bands, as shown for two conjugated polymers P3HT and PCPDTBT in comparison to CuInSe2. This drawback could be circumvented by synthesis of novel materials, or multijunction concepts (tandem solar cells).
Continue reading “Picture Story – How Do Organic Solar Cells Function?”




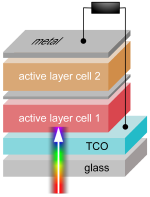

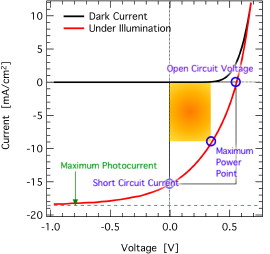 made for crystalline inorganic diodes, when applied on fitting organic solar cells.
made for crystalline inorganic diodes, when applied on fitting organic solar cells.
