As I won a proposal today, I feel up to contributing once again some physics to this blog… I know, it has been a long long wait. So today it is time to consider some fundamentals of charge transport, as this is not only important for the extraction of charge carriers from the device  (see earlier posts on mobility and efficiency, surface recombination velocity and photocurrent) but also the nongeminate recombination (see e.g. photocurrent part 2 and 3).
(see earlier posts on mobility and efficiency, surface recombination velocity and photocurrent) but also the nongeminate recombination (see e.g. photocurrent part 2 and 3).
In disordered systems without long range order – such as an organic semiconductor which is processed into a thin film by sin coating – in which charge carriers are localised on different molecular sites, charge transport occurs by a hopping process. Due to the disorder, you can imagine that adjacent molecules are differently aligned and have varying distances across the device. Then, the charge carriers can only move by a combination of tunneling to cover the distance, and thermal activation to jump up in energy. In the 1950s, Rudolph A. Marcus proposed a hopping rate (jumps per second), which is suitable to describe the local charge transport. By the way, he received the 1992 Nobel prize in chemistry for his contributions to this theory of electron transfer reactions in chemical systems.The equation he proposed for the hopping rate from site i to site j across the distance  is
is
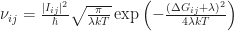 .
.
Here, 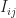 is the transfer integral, i.e. the wavefunction overlap between sites i and j, which is proportional to the tunnelling contribution.
is the transfer integral, i.e. the wavefunction overlap between sites i and j, which is proportional to the tunnelling contribution. 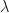 is the reorganisation energy related to the polaron relaxation, which is sometimes called self-trapping: the molecule is distorted by the charge, which leads to a (lattice) polarisation, lowering the site energy.
is the reorganisation energy related to the polaron relaxation, which is sometimes called self-trapping: the molecule is distorted by the charge, which leads to a (lattice) polarisation, lowering the site energy.  is the thermal energy and
is the thermal energy and 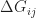 is due to different energetic contributions, in particular the energy difference between the two sites. In disordered systems, the density of states is often approximated by an exponential or Gaussian distribution, so that the energy of each site is from this distribution. Integrating over all site energies just yields the chosen energy distribution, e.g. a Gaussian, once again. Then, is just the energy difference of the two chosen sites. Thus, jumping from one molecular site to the next is proportional to the tunneling term and an exponential term proportional to the site energy difference and the self-trapping of the charge on the initial molecular site.
is due to different energetic contributions, in particular the energy difference between the two sites. In disordered systems, the density of states is often approximated by an exponential or Gaussian distribution, so that the energy of each site is from this distribution. Integrating over all site energies just yields the chosen energy distribution, e.g. a Gaussian, once again. Then, is just the energy difference of the two chosen sites. Thus, jumping from one molecular site to the next is proportional to the tunneling term and an exponential term proportional to the site energy difference and the self-trapping of the charge on the initial molecular site.
For a given molecule, the arrangement can be calculated by molecular dynamics, and the transfer integrals between different possible pairs of molecules, constituting sites i and j, respectively, can be calculated by quantum chemistry. A nice application of this approach is shown in [Kirkpatrick 2007] for discotic liquid crystals, without considering molecular dynamics in a qualitative way look at [Stehr 2011].
A simpler but more generic way to calculate a hopping rate is the so-called Miller-Abrahams hopping rate
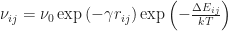 .
.
Here, the contributions of tunnelling and thermal activation are even more explicit. 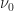 is the maximum hopping rate, sometimes called attempt-to-escape frequency.
is the maximum hopping rate, sometimes called attempt-to-escape frequency. 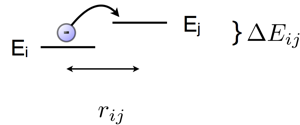
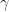 is the inverse localisation radius, stating how well charge carriers can tunnel across the distance between site i and j. Indeed, the first term denotes the tunneling contribution. The thermal activation comes from a Boltzmann term, where hopping upwards in energy, i.e.
is the inverse localisation radius, stating how well charge carriers can tunnel across the distance between site i and j. Indeed, the first term denotes the tunneling contribution. The thermal activation comes from a Boltzmann term, where hopping upwards in energy, i.e.  >0: if the hopping process is from an initial state i lower in energy than the final state j, it is made difficult by an exponential penalty. Hopping downwards in energy (<0) is approximated to be always similarly easy: the complete second term, the Boltzmann term, is replaced by
>0: if the hopping process is from an initial state i lower in energy than the final state j, it is made difficult by an exponential penalty. Hopping downwards in energy (<0) is approximated to be always similarly easy: the complete second term, the Boltzmann term, is replaced by  . In the Miller-Abrahams rate, the molecular details are usually neglected, so instead of transfer integrals only the attempt-to-escape frequency is approximated. Instead of the reorganisation energy, only energetic site differences derived from a (often Gaussian) density of states distribution are considered.
. In the Miller-Abrahams rate, the molecular details are usually neglected, so instead of transfer integrals only the attempt-to-escape frequency is approximated. Instead of the reorganisation energy, only energetic site differences derived from a (often Gaussian) density of states distribution are considered.
Both models, Marcus and Miller-Abrahams hopping rate, are used in different context and are not exactly equivalent, but will yield similar results under many conditions. Nevertheless, it is probably safe to state that the former has a higher scientific applicability.
Now why is it important to be able to calculate a hopping rate when considering charge transport in organic matter? – If one knows the number of molecular sites across the device length 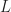 , which is the conservative estimate of the number of jumps
, which is the conservative estimate of the number of jumps 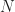 needed to travel through the whole device, and the time needed per jump
needed to travel through the whole device, and the time needed per jump 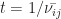 , one can calculate the velocity
, one can calculate the velocity 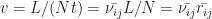 . Here,
. Here,  is the average distance crossed per single jump. If the velocity is known, also the charge carrier mobility is known, which is a very important figure of merit in semiconductor physics. The mobility
is the average distance crossed per single jump. If the velocity is known, also the charge carrier mobility is known, which is a very important figure of merit in semiconductor physics. The mobility  relates the drift velocity
relates the drift velocity  to its driving force, the electric field
to its driving force, the electric field 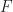 , so that
, so that 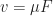 . A lot of essential information on charge transport is included in this inconspicuous parameter , especially if disorder is considered.
. A lot of essential information on charge transport is included in this inconspicuous parameter , especially if disorder is considered.
The charge carrier velocity can, thus, be calculated by knowledge of the hopping rate as well as the time for each hop and the number of hops. Also, can alternatively be determined experimentally by measuring the transit time of charge carriers through a device of known thickness. Thus, a direct comparison of experiment and simulation is possible and desired for grasping how charge transport works. A suitable and very straight forward experiment is the transient photocurrent, also called time-of-flight (TOF) measurement. A fitting computer model is based on a kinetic Monte Carlo simulation, in which a certain spatial and energetic distribution of sites is assumed and the Marcus or Miller-Abrahams hopping rates are calculated.
Next time, I will explain the TOF experiment, and then Monte Carlo simulations. Both together allowed (and still alow) to much better understand charge transport in disordered organic semiconductors.
Add to Connotea


Great article! You mentioned that the Miller-Abrahams model is not necessarily appropriate for modelling the same things as the Marcus model. Could you give an example of when the Miller-Abrahams model would be a more accurate representation of a system. I understand that the Marcus model takes into account polaron effects and the Miller-Abrahams doesn’t explicitly account for them.
Sorry, forgot to answer… and happy new year, by the way!
If you compare the rates for Miller-Abrahams and Marcus hopping rates, they are pretty similar. Tunnel term: attempt-to-escape frequency in MA contains Marcus’ transfer integral. Boltzmann term: you are right, Marcus contains the reorganisation energy instead of disorder. However, you can modify the activation energy in MA to consider disorder and/or polaron effects as well. The main difference is that Marcus theory has an inverted regime where, for hops far down, the hopping rate decreases again. In contrast, for MA, all hops downwards in energy have the same maximum hopping rate.
Hi again, I realize that the inverted regime that you mentioned has been supported by experimental data, but has anyone investigated the mechanisms responsible for it? Is the inverted regime a result of competing processes?
Thanks for sharing your knowledge.
Hi again , the inverted regime was predicted by R. A. Marcus himself, and later proven experimentally. I just checked has Nobel prize lecture in paperform [Marcus 1993], around Fig. 6 and 7 you find the explanation which does not require additional competing processes. Sorry for not going into detail, but dinner with family waits;-) Best, Carsten
, the inverted regime was predicted by R. A. Marcus himself, and later proven experimentally. I just checked has Nobel prize lecture in paperform [Marcus 1993], around Fig. 6 and 7 you find the explanation which does not require additional competing processes. Sorry for not going into detail, but dinner with family waits;-) Best, Carsten
Thanks, for your comments. I was unable to find a satisfactory description in Marcus’s 1993 paper, but one of the papers he references by J. R. Miller does provide a reasonable if somewhat brief explanation.
http://pubs.acs.org/doi/pdf/10.1021/ja00322a058
I believe they are suggesting that the inverted regime is caused from an increased mismatch in the overlap of the vibrational wave functions.
Hey, I can’t seem to find your continuous post on Monte Carlo. I am curious to read on :)
Yes, hmmm, plans vs reality…
I fear there are some more promises on this blog which deserve to be fulfilled. But thanks for pointing this one out, I really should do at least the “cartoon version”;-)