
I’ve talked a lot about polaron pairs and excitons lately, and will continue to do so,  that this time I’ll give short explanations of what I am actually talking about. Call it definitions… ;-)
that this time I’ll give short explanations of what I am actually talking about. Call it definitions… ;-)
A polaron is a charge, i.e., an electron or a hole, plus a distortion of the charge’s surroundings. In a crystalline inorganic material, setting a charge onto a site does not change the surroundings, as the crystal lattice is rigid. Not so in many disordered organic materials. Putting a charge onto a certain molecular site can deform the whole molecule. Moving the charge from this to another molecule means that first the energy for the deformation – the polaron binding energy or reorganisation energy – has to be mustered. The implication is that charge transport becomes more difficult, the charge carrier mobility becomes lower, … This process is also described as self-trapping. As a side note, it is often difficult to distinguish between the influence of polaronic self-trapping and of gaussian disorder, as both have a similar impact on the charge transport properties. This similarity is also reflected in the corresponding hopping rates used to calculate charge transport: Marcus theory is a function of the reorganisation energy, where as the Miller Abrahams rate [Miller 1960] is related to the energetic disorder of the density of states. The polaronic deformation can be quantified in terms of a (lattice) polarisation, or a phonon cloud, or just as the above-mentioned polaron binding energy. Mostly, however, when hearing polaron, think charge;-) See also what wikipedia has to say about polarons.
A polaron pair is a Coulomb bound pair of a negative and a positive polarons, situated on different molecules. Usually, polaron pairs are the intermediate step from an exciton to a pair of free polarons &ndash far enough apart not to feel the attraction of one another &ndash and therefore important in order to understand photogeneration in organic semiconductors.
An exciton is an excited quasiparticle in a solid, which is formed by a Coulomb-bound electron-hole pair. It is more prominent in organic semiconductors as compared to their inorganic counterparts: as the dielectric constant is lower in organics, the screening length is larger. In this case, the name Frenkel exciton is applied, whereas the weakly bound type is called Wannier-Mott. Thus, in organic materials, the two charges feel a strong mutual attraction, and usually reside on one molecule. There seem to be special cases, however, in which the two particles reside on adjacent molecules – of the same kind, in contrast to polaron pairs. The spin-state of the two charges is quite important. Without going into too much detail: when the two spin-vectors add up to zero, we have a singlet exciton. Singlet excitons are the only ones which are generated upon illumination, which is due to the specific selection rules. The other exciton type, triplet excitons, have a nonzero spin vector, which is possible in three different combinations – thus the name triplet. Singlet and triplet excitons can also be formed due to interaction following charge injection; theoretically, this follows a one-to-three ratio, i.e., only a quarter is of singlet type. Some features of singlet excitons and their relevance for organic photovoltaics was discussed here. The exciton binding energy of singlets is around 0.3eV in organics (compared to ~0.01eV in classical semiconductors). Excitons have a certain lifetime, typically of the order of ns in organic semiconductors, after which they recombine radiatively; this is called photoluminescence. Triplet excitons generally have lower energies and longer lifetimes. For photovoltaics, they are not yet import (though might be following some novel concepts),  instead they can act as loss mechanisms (by intersystem crossing or electran back transfer) under certain conditions as their energy is too low to generate free charge carriers. Radiative recombination after the triplet long lifetime of maybe some milliseconds – the transition is actually spin forbidden – phosphorescence occurs. As a side note, phosphorescence can be applied to high usefulness in so called triplet emitters, being an important concept for organic light emitting diodes. Maybe we’ll detail this another time. Wikipedia on excitons here.
instead they can act as loss mechanisms (by intersystem crossing or electran back transfer) under certain conditions as their energy is too low to generate free charge carriers. Radiative recombination after the triplet long lifetime of maybe some milliseconds – the transition is actually spin forbidden – phosphorescence occurs. As a side note, phosphorescence can be applied to high usefulness in so called triplet emitters, being an important concept for organic light emitting diodes. Maybe we’ll detail this another time. Wikipedia on excitons here.
An exciplex is just an exciton which is located at the interface of its “host” molecular material – indeed it still resides on one molecule – as indicated in the image. Due to the influence of the surface, the exciplex experiences a different environment as compared to a bulk exciton. This leads to photoluminescence which is slighlty red shifted. Also, the lifetime can be prolonged in comparison to the bulk exciton, as it is stabilised by the surface states.
Did you miss bipolarons? I didn’t;-)
Thanks to JG for the exciplex!
[Update 27.4.2010 to answer the question of Jenna] In organic bulk heterojunction solar cells, the path from singlet excitons in P3HT to free charges usually goes via charge transfer complexes of the donor-acceptor system. (See for instance here.) I often refer to these as polaron pairs. However, naming conventions are not that simple. Here a brief excerpt from an unpublished review I recently wrote (accepted for publication by Adv Mater).
The commonly used names for CT states and complexes are diverse, either used alternatively or to define special cases. Examples are polaron pairs, [Dyakonov 1998] intermolecular radical pairs (with the radical cation on the polymer and the radical anion on the fullerene) [ Scharber2003], interfacial charge pairs [Westenhoff 2008], geminate pairs [Arkhipov2003], charge transfer excitons [Veldman2008] and exciplexes [Morteani2004].
Huang et al. [Huang 2008] found by theoretical considerations for polymer-polymer heterojunctions that a range of Coulombically bound CT states with both, emissive and non-emissive character, exist. The different states are a result of the specific features of the intermolecular overlap between donor and acceptor moieties. In order to strive for a more precise nomenclature, they point out that polaron pairs can be considered as one special instance of the more general exciplex. From this point of view, the distincitve property of the polaron pair excitation is that it is due to a complete charge transfer from donor to acceptor, as opposed to a partial CT. Thus, an exciplex can generally be regarded as a hybrid state with partly CT character and a certain fraction of a local excitation on one (or both) molecules of the donor–acceptor system. Already earlier, Gould et al. [Gould 1994] pointed out that the character of the emitting species of an exciplex depends on the relative contributions of pure ion-pair and locally excited states. In their definition, an exciplex with beyond 90% CT character represents a pure contact radical-ion pair. They suggested that it can be identified experimentally by verifying that the emission maximum lies about 5000/cm (100meV) below the singlet exciton photoluminescence.

Hello,
Firstly, thank you for your blog! I’m finding it very useful as I’ve just started a PhD in organic solar cells.
I have a question on exciton-types. Hopefully you might be able to shed some light on my confusion. I’m looking at systems in which Frenkel and Charge-Transfer exctions have been found but I’m trying to get my head round exactly what a CT exciton is. In my (maybe simplified view) it was simply an exciton created when the electron from one material (e.g. polymer) was promoted to (close to) the LUMO level of another material (e.g. fullerene). The article below mentions them but in context of CT states being filled by Frenkel excitons.
With regards to your diagram would the CT exciton be equivalent to the polaron pair? I presumed it wasn’t or else they’d call it a polaron! It is definitely something shared between materials and no on one molecule so it isn’t an exciplex. Can the polaron pair be generated directly by light absorption?
http://www3.interscience.wiley.com/cgi-bin/fulltext/122630062/PDFSTART
If you can clarify this at all I’ll be very grateful!
Jenna
Hi Jenna, thanks for your kind comment! Concerning your question, I have added a hopefully helpful update at the end of the post. Best, Carsten
Hello,
I am really anxious to know that what will happen when two polarons are forced in proximity of each other?? will they make an exciton polaron pair??
Anticipating your quick response.
Thanking in advance.
In a single material, two approaching charges can come together to form an exciton, and this would be a downhill in energy process, although recombination directly is more likely as this is even more downhill. In a bilayer solar cell please read some of the papers from Cambridge University on the possibility of reforming an exciton from two charges. There is an energy barrier at the interface that makes it unlikely but it seems not impossible.
Hi Carsten, thanks so much for your prompt and full reply! It has helped me to think about the range of states possible and what they physically mean. Best wishes, Jenna.
We do need some definition for terms, and the description you offer here is a good start but I dont know if i would describe them exactly as you do here. Firstly your description of an exciplex is not great; an exciplex is an excited state complex between two different chromophores. By definition the excited state is shared by the two chromophores. If the two chromophores are the same you get an excimer (excited state dimer). You also dont differentiate between chromophores and molecules. Also there is a problem with what is the actual difference between your definition of an exciton (a bound electron hole pair) and a polaron pair (a bound electron hole pair). For me, possibly the easiest definition of the difference is that in an exciton , hole and electron effect their surroundings as a unit, i.e. they relax as a unit. In a polaron pair , there are two polarons which relax individually into their surroundings, but due to their proximity apply coulombic interaction on each other. But this is only what i think. As a community we definitely need agreed upon definitions and we need them soon.
Hi Dr. Deibel,
Thank you so much for posting so many important terms in your blog, which is really helpful.
I have a question to ask here about the excitons generation due to charge injection. In this page, as well as in your paper (Rep. Prog. Phys. 73 (2010) 096401), you mentioned “Singlet and triplet excitons can also be formed due to interaction following charge injection”. I’m really curious about how this process happens, because it’s very important to understand the carrier transport at equilibrium and steady state.
Thank you so much and look forward to your reply.
Kejia
Hi Kejia, thanks for your question: it is a good one, and would deserve a post, but I’ll try to give ou a starting point. Singlet and triplet generation upon injection is more relevant for charge injection based devices such as organic light emitting diodes. These are made of a single organic semiconductor, not a blend. Thus, the lowest excited states are “normal” excitons, not charge transfer excitons (or polaron pairs) across a donor-acceptor heterojunction. As the spins of the two exciton constituents can each be up or down, four configurations are possible – one singlet and three triplets. If spin statistics work, you will have a 25% chance of forming a singlet exciton upon injection an electron and an (uncorrelated) hole into the organic semiconductor. (If the chance can be increased beyond 25% is an important question for the quantum efficiency of OLEDs; interesting albeit older reviews are [Yersin 2004] and [Wohlgenannt 2003], also including the detailed mechanisms). For solar cells, the optical excitation is more important: optically, usually only singlet excitons (for instance on the donor material) are generated, which can become triplets directly only by intersystem crossing. The latter can be on a picosecond time scale, but is still slower than the fs electron transfer, and thus less favourable. Another triplet generation mechanism is electron back transfer, which can occur after successful electron transfer if the charges do not find enough transport paths to get away from the interface (and if the process is energetically possible). For some further reading, see [Veldman 2009].
Hi, Dr. Deibel , thanks a lot for your post. I am a graduate student, have a question about polarons. Does polaron or polaron pairs have the temperature dependence?
Your question is not clear to me, sorry.
Hi, my question is regarding solitons.Can polyaniline have solitons as charge carriers?Is it correct that only conducting polymers like polyacetylene can have solitons as charge carriers?
I never cared much for solitons, sorry. Have you checked the book by Pope/Swenberg? C
Thinking that the topic of exciton splitting is of increasing interest but without any concrete definitions, I thought I would offer some interpretations of the ‘meaning’ of states. Looking forward to some comments.
Definitions:
“geminate charge pair” – hole and electron charge created by the same individual event
‘to separate’ – charges increase their physical separation distance
‘to dissociate’ – the presence of the nearby opposite charge no longer biases charge movement direction – the charge is ‘free’
‘to approach’ – charges decrease their physical separation distance
‘to associate/capture’ – when the presence of a nearby opposite charge starts to bias charge movement direction –
‘correlated’ – hole and electron act as a single unit so that energy transitions involving energy moving in and out of the pair cannot be discerned as coming or going from either hole or electron components. Examples of these would be neutral excitons or exciplexes. As there needs to be a lot of electronic orbital overlap for this to occur, hole and electron would normally be confined to much less than 20A.
‘relaxed (geminate) charge pair’ or ‘relaxed radical ion pair’ – (geminate) charges at the smallest possible separation distance so that coulombic potential is at a minimum (coulombic attraction is maximised) , and with no excess vibrational or electronic energy. They are however not correlated and act as individual units. In this sense, ‘exciplexes’ and ’relaxed charge (ion) pairs’ are opposite ends of a spectrum, with exciplexes showing almost no charge transfer character (nearly perfectly neutral) while ‘relaxed charge (ion) pairs’ show complete charge transfer.
‘unrelaxed charge pair’ – are not at the minimum possible separation distance physically or have excess vibrational or electronic energy
Thank you, Carsten, for an excellent “definition” post, and thanks to others for thier excellent comments.
Can someone point me to a treatment of excitons, polarons and related, in disordered solids under large electric field? I am specifically thinking of amorphous metal oxides, but any material is OK. By high E-field, I mean ~ 1 MV/cm = 100 MV/m = 0.1 v/nm and somewhat higher.
I am new in this blog/forum, so I am guessing that most of you are working with solar cells or photovoltaics, in which there may be a mild “drift” E-field, to sweep separated charges to their respective collection electrodes. What magnitudes are those fields, typically?
The operating voltage of some organic solar cells is 1V over a 100 nm thickness so I guess drift electric fields of up to 0.1MV/cm is common. You need to go higher before you start to distort excitons, or polaron trap states significantly in organics…although I don’t know much about metal oxides.
Cheers. Consider the thickness of the device, which is 100 to 300nm for state-of-the-art solar cells. The built in potential of organic solar cells is usually 1V or less, and the open circuit voltage is somewhat lower. The solar cell supplies power to the external circuit in the 4th quadrant of the IV curve, thus between short circuit and open circuit conditions, with the highest power at the maximum power point… which again is usually lightly below Voc. Thus, 1V/100nm is the upper limit for the internal field under operating conditions, with a typical range of, say,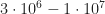 V/m. Therefore, field dependent exciton dissociation does not occur in this regime, only generation, energy relaxation (during diffusion), electron transfer to the polymer (or hole transfer from fullerene to polymer) and, about 1ns after generation, radiative or nonradiative decay of the excitons which did not undergo charge transfer.
V/m. Therefore, field dependent exciton dissociation does not occur in this regime, only generation, energy relaxation (during diffusion), electron transfer to the polymer (or hole transfer from fullerene to polymer) and, about 1ns after generation, radiative or nonradiative decay of the excitons which did not undergo charge transfer.
Why it is said like solar cells supply power to the external circuit in the 4th quadrant of the I-V curve?
It is a question of definition. Here, in the 4th quadrant at maximum power point, the voltage is positive and the photocurrent negative = photocurrent comes out of the device. The product of voltage and current is also negative: power delivered from the device to the circuit.
Thanks, inever…jack. (too lazy to type your full screen-name)
So 1/10th or so my working field does not distort exitons, and that field is common in organics. I think the dielectric constant of metal oxides can be significantly greater than that in organics, and with that comes greater static polarization, both electronic and atom-core, of the medium and more effective screening of separated charges. I wonder how larger field and greater polarizability work together to affect excitons, polarons, etc.? Maybe I do need a teaching that is specific to metal oxides, though I am not working with extremely high k values, only 10 to 40.
Dr. Deibel, one concept that I have often seen but never quite grasped when comparisons are made between excitons in inorganic and organic semiconductors is that of “screening”. In your post you state: “the dielectric constant is lower in organics, the screening length is larger.” What does this mean?
Talha,
At a simple level, dielectric constant measures the polarizability of (let’s limit to) the electrons in a solid. Polarizability (dipole moment per unit volume [m*C/m^3] = [C/m^2]) expresses how much the electron density distorts in reponse to an electric field other than that of the native nuclei and other electrons of the solid. (Of course, there is already native, local polarization various places in the solid, for example, C-O bond polarization.) Screening length (similar to Debye length in a plasma) measures how far away from a non-native charge (a so-called “test charge”) you would have to be in order to be completely unable to detect its presence. In a non-polarizable material, the distance would theoretically be infinity. In a highly polarizable material, the native charges will rearrange (distort their density distributions) to lower their potential energy in the electric field of the test charge, thereby crudely cancelling the far field of the test charge. The more polarizable the medium, the more readily and more effectively the native charges move to do this. Therefore the far field of the test charge is reduced. When the far field of the test charge is reduced to ~ the same magnitude as the space-charge fluctuations in the native electron density, we say that the test charge is “screened”. Whatever distance away from the test charge this occurs is called the “screening distance”. Therefore, higher dielectric constant –> more polarizability –> easier charge distortion –> shorter screening distance. All clear? (Other Members, please correct me, for Talha’s sake, if I mis-stated the situation.)
Hi Talha, hi David (and thanks for the contribution concerning the dielectric constant!)!
An excitation in an organic semiconductor, e.g. by absorbing a photon, can be (very roughly!) compared to a strongly bound electron-hole pair with a certain interpair distance. If you consider this distance and the dielectric constant of the material, you can calculate the Coulomb binding energy, which for dielectric constant and a radius of 1nm comes out at about half an electron Volt. David nicely explained the screening (and see also this Wikipedia entry for screening and Coulomb interaction), so
and a radius of 1nm comes out at about half an electron Volt. David nicely explained the screening (and see also this Wikipedia entry for screening and Coulomb interaction), so
lowhigh screening length comes forhighlow dielectric constant and implies a strong binding energy. [Update 2018-12-26 fixed mistake and fixed the fix. Thanks Calvin and David!]This means if you absorb a photon at the absorption edge of 2eV, and an exciton is generated (here with 0.5 eV binding energy), you need an additional 0.5 eV energy to convert this exciton into independent (=not interacting with one another) charge carriers. As at room temperature, your thermal energy is only 1/20 of the exciton binding energy, it is not sufficient to dissociate the exciton… leaving, e.g., the electric field as driving force (and, of course, the electron accepting fullerene in a blend system). Charge extraction experiments and PL quenching (see [Hertel 2002] and others) show that for neat polymers, you need a high electric field (say,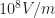 ) in order to extract charge carriers after a short laser pulse. However, some people argue that the exciton binding energy in these materials is not the limiting step — which inever…jack can probably explain better (not that I introduce so many errors in my text when talking about ultrafast photophysics experiments, eh;-)
) in order to extract charge carriers after a short laser pulse. However, some people argue that the exciton binding energy in these materials is not the limiting step — which inever…jack can probably explain better (not that I introduce so many errors in my text when talking about ultrafast photophysics experiments, eh;-)
Best, Carsten
This blog really helps. Thank you very much.
I am not sure if people is still replying, but I am going to ask anyway.
I found something that is not consistent.
In the article, Dr. Deibel said “the dielectric constant is lower in organics, the screening length is larger”. I know that organic semiconductor exhibit low-k and has strong coulomb interaction .
But then in the comment he said “low screening length comes for high dielectric constant and implies a strong binding energy.”
His comment make me confuse. Does organic semiconductor exhibit high-K or Low-K?
Calvin MS,
First, the answer to your last question: Organic semiconductors have low k.
Here are the correct trends:
Lower dielectric constant –> longer screening length and/or stronger coulomb interaction.
Higher dielectric constant –> Shorter screening length and/or weaker coulomb interaction.
The basic formula is the one for energy between two point-charges in a vacuum. Even though the charge distributions are diffuse at the size-scale of excitons, polarons, etc. and the environment is not a vacuum, the concepts and trends are the same.
For two charges q1 and q2 separated by a distance r12, in a medium with dielectric constant k, so permittivity is ε = kε0, the coulomb electrostatic energy, Ecoul, is:
Ecoul = q1q2/(4πεr12)
This equation is the origin of the two trends stated above. You can see immediately that when ε is larger, Ecoul is smaller and vise-versa, at a fixed distance r12. You can get an idea about screening length by rearranging the equation:
r12 = q1q2/(4πεEcoul)
and setting the energy Ecoul to kBT, where kB is Boltmann’s constant, which means kBT is approximately the thermal energy. Then the distance r12 is approximately the screening length (this is not a rigorous derivation). Then you can see that when ε is larger, r12 is smaller and vise-versa.
With all that said, yes, I think you did catch an inconsistency. They are just some typos, but in his comment on 19 May 2012 at 9:09, deibel said:
“low screening length comes for high dielectric constant and implies a strong binding energy.”
Then in his Update 2018-12-26 fixed mistake. Thanks Calvin!, deibel said:
“low screening length comes for low dielectric constant and implies a strong binding energy.”
They are both wrong. More correct would be:
“low screening length comes for high dielectric constant and implies a weak binding energy.”
It is a 3-way relationship, and I am sure that our master deibel just got in a hurry and alternately got each of the 2-way relationships correct.
Thanks David!
Calvin MS,
I refine my own comment. In a strong, rigid solid (even if amorphous, as is the case for dense amorphous metal oxides), the ε in the formula
Ecoul = q1q2/(4πεr12)
should represent only the electronic component of the polarization, not the nuclear movement components (partial rotations, vibrational distortions, shift in average position of net-charged ion cores, etc.). So my relation for permittivity as ε = kε0 is not right, if we use the full value of k.
We need to use this electronic component of the polarization if the charge, e.g., a negative polaron, is moving. Electronic motions are so fast relative to ionic and nuclear motions that there is no time for heavy-particle polarization motions – the electron will be gone before the nuclei can move.
Whoops; I made an error. I said “Polarizability (dipole moment per unit volume [m*C/m^3] = [C/m^2])….”. In fact, it is polarization that is dipole moment per unit volume and has those units.
Polarizability would be dipole moment induced by a certain applied electric field. So polarizability would have units [C*m]/[V/m] = [(C*m^2)/V], where V is “volt”. Is this correct?
I guess is would also be meaningful to define polarizability as polarization induced by a certain applied electric field. In that case, polarizability would have units [C/m^2]/[V/m] = [Farad/m], so some measure of reduced capacitance.
Anyway, whichever way you think of polarizability, greater polarizability expresses easier charge density distortion per unit of applied electric field. The remainder of my explanation of screening length should be OK.
Hi Carsten
I am Joydeep , have a quick question. In a paper I found that only 1% of the whole exciton population recombine radiatively giving off photo luminescence . Others are recombined non radiatively. Could you please put some light on non radiative recombination pathways of exciton in organic solar cell.
Hi Joydeep, you probably refer to [Piris 2009] on P3HT. Others put the number of radiative recombination to maybe 10% for this material (do not have reference at hand, sorry). A major reason is probably the H-aggregate nature of P3HT. Instead of a more detailed answer (to which inever…jack would be more suited anyway;-), I refer you to the real specialist;-) Have a look at papers by F. C. Spano, for instance [Spano 2005]. Hope that helps or at least gives you a good start for looking deeper, best, Carsten
Thanks Carsten
I wish I knew what the real number of radiatively recombining excitons was, in photochemistry it is one of those values that is hard to measure right, it can take decades and numbers are always being revised upwards. There are several issues with measurements, none of them new, including wavelength dependence, avoiding singlet singlet annihilation and avoiding degradation of sample, the last being crucial for an accurate measurement. Definitely closer to 5% radiative efficiency for P3HT but could be more than this in reality, perhaps as far as the 20-30% seen in other polymers.
Non-radiative reasons for exciton decay. Well; conversion from singlet to the less fluorescent triplet, multiphonon dissipation of energy either by the chromophore itself or with the help of an impurity. Conversion of the singlet to an exciplex or excimer. Energy transfer (Forster or Dexter) to an impurity. Natural blinking or due to energy migration to a site that is currently blinking (read single molecule spectroscopy papers to understand this). Charge transfer against another chromophore (exciton splitting) or charge transfer against an impurity. Singlet-singlet annihilation, singlet-triplet annihilation, singlet-polaron annihilation.
But in a solar cell hopefully the overwhelmingly means of non-radiative recombination will be exciton splitting (charge transfer) and all other processes will be slow compared to this.
I have a “stupid question” about excitons. Exciton spin states are either singlets or triplets. I understand that for electron pairs like cooper pairs but I do not understand how can a hole have spin. Please help.
As much as a hole is “only” a missing negative charge within a band full of electrons, the hole has a “missing” (and unpaired) spin.
It is not that the hole has a spin, it is that the electron that was left behind when one of the electrons in a pair was promoted, has the spin.
I did say unpaired… nitpicker (at least one of us;-)
Hey Carsten,
I am trying to understand the Onsager-Braun Model which gives the dissociation rate of polarons. I don’t get what is meant by “thermalisation radius” (in your paper “advanced materials 2010, 22, page 4101” you say its “the distance at which the polarons of the initial CT complex relax thermally”). Could you describe it in a bit more detail? Is “relax” = “recombine”? And how does a radius determine the way how polarons relax? Can you predict if the relaxation of polarons is thermally or e.g. radiatively or by emitting phonons? And how is this radius determined by material properties? Is ther thermalisation radius different for a PCBM/P3HT compared to a ZnO/P3HT system?
Cheers for reply!
Miriam
Hi, this is a difficult question but maybe i will have a go. Onsager was a brilliant scientist and mathematician and he wrote an expression that more realistically simulated the movement of ions in a solution, a solution that took into account among other things the coulomb attractive force between ions and the effect of external fields. Now, one thing Onsager’s equation didn’t have is a time related part, as he never needed it, as there would always be a steady state concentration of ions in solution. At this point Braun modifies Onsager’s theory to introduce a time part to the ion concentration, e.g., if a positive and negative ion were close together for long enough they would eventually recombine and both ions vanish. This Onsager-Braun theory is a useful theory for looking at the recombination of positive and negative electrical charges in solutions and also perhaps more controversially solids. By Onsager theory there is a distance where the coulombic attraction of opposing ions overwhelms their normal motion and the charges will inevitably come together and recombine. This is called the Onsager radius (or sometimes capture radius).
You asked about the thermalisation radius. Well, the thermalisation radius is important because; some time (<50 fs I guess, but not instantaneous by any means) after the formation of a geminate pair they will settle to a finite distance apart, if that thermalisation distance is greater than the onsager radius then they will escape each other's attraction, if their thermalisation distance is less than the onsager radius then they will almost definitely recombine. So what controls the thermalisation distance? Well, the initial distance of electron transfer is probably a part of the thermalisation distance but this is usually less than 2 nm. More importantly, after geminate pair formation the charges may (or may not) have excess energy, and this excess energy as it is lost to the surrounds leads to the electron and hole becoming further separated (despite the coulombic attraction – in fact the coulombic attraction may be one way of how that energy is lost quicker). This is a bit like a phonon-electron scattering process. Once the charges are at an equivalent energy to their surroundings (they have no more energy to give away) they will stop being driven apart and will have thermalised at the thermalisation radius. You would think that as the thermalisation process is a diffusional process it should depend highly on the system i.e. on morphology, mobility and other factors like dielectric constant. Therefore P3HT:ZnO and P3HT:PCBM should behave differently (but then again maybe not!). Unfortunately, there has been not much work done on this field in the last two decades, due to it being very dry and difficult and maybe it is only recently with organic semiconductors becoming important that work on this has restarted. What work there has been in the past must now be re-evaluated given the ever better equipment at our disposal and hopefully this matter of geminate pair dissociation after exciton splitting can be modelled better. I hope this helps.
Very nice reply! Although implicit in inever…’s answer, for many materials the Braun-Onsager model cannot be used to fit the photogeneration properly, as experimental field and (in particular) dependence are weaker. If a hot process during the generation contributes, indeed do not know enough to adjust parameters such as the “thermalisation radius” in order to include these processes. As an additional note, the Braun-Onsager equation considers the Langevin recombination for describing the recombination back from free to bound state. The “thermalisation radius” is in this case used to calculate a virtual charge carrier density (1/Volume). Details on demand, need to pick up my wife from the train;-)
Dear Dr. Deibel,
I saw in one of the posts of “Ineverwanted….. ” that he raises the question of the differences between what is meant by a polaron pair and an exciton.I could not find the answer.Can you please shed some light on what actually are the differences between a exciton, a polaron pair, and an electron hole pair..
Thank you!
Hi! The difference between the electron-hole pair and a polaron pair (which also is made of one electron and one hole) is mainly that even single polarons are electrons which distort their surroundings (e.g. in organic semiconductors) as opposed to electrons in, e.g., an inorganic crystalline solid. Both usually are Coulomb bound, but the electron-hole pairs in inorganics are usually bound much more weakly. For excitons it is more complicated. Originally, I wrote “An exciton is an excited quasiparticle in a solid, which is formed by a Coulomb-bound electron-hole pair.”. However, this is only correct in a single-electron approximation. It is more general, and for organics probably more accurate, to talk about neutral (photo)excitations of a molecule. Compared to the unexcited molecule, the binding energy of the exciton is significant. Using very high electric fields or a second material with suitable acceptor to overcome the binding energy, the exciton can be dissociated into polaron pairs or even free (unbound) polarons. As your question probably goes deeper, please keep on asking. While I am not “Inever…” I will try to answer (or tell you if I do not know;-) Cheers, Carsten
Dear Dr. Deibel,
The information in this page is very helpful to understand the fundamental of light induced photophysical processes in OPVs. In one of the paper I came to know about delocalised polaron pair. Can you please tell me the diffrerence between localised and delocalised polaron pair…
Delocalised and localised (polaron pairs) are really ambiguous definitions. Consider them more as two ends of a scale then as black and white definitions. A (very) localised charge (half a polaron pair) would exist only inside a single orbital of an atom. An example of this would be gas phase experiments where atoms pick up charges; the charge is then localised only on that atom. A delocalised charge is where the charge is shared between many different atoms and effectively spreads itself out over a large distance. This in reality is the case for organic semiconductors and other condensed matter semiconductors, the charges are spread out between several atoms and have an averaged appearance. Delocalisation occurs because in the overwhelming majority of cases sharing the charge lowers the overall energy state of the material holding that charge. In many papers when authors discuss delocalised/localised polarons all they mean is that some charges are more spread out than others.
I agree with inever… It seems to me that it is often difficult to make the assignment localised very delocalised experimentally. I think [Österbacka 2000] made their assignment by comparing to neat polymer dispersed in polystyrol or so. In that paper, however, it looks more like the “black and white” picture. I wonder if the polaron signature in photoinduced absorption always shows a (continuous) shift proportional to the degree of delocalisation. Best, Carsten
how the mechanism of organic solar cell work is explained very well.thank you for such explanation.
can you give the explanation of hopping transport in organic solar cell and also the localized and localized state with figure.
I think Carsten has already done that in many of his previous posts. Please have a look. Do you have anything in particular you want to discuss?
Back to Palas Roy’s question of delocalization, I have a thought [amazing that this thread goes on for years now!].
Is it not true, for a negative polaron (let’s accept that case for now) exhibiting a tendency to delocalization of the extra electron, that
a) the externally applied E-field would spatially distort the “orbital” (one-electron wavefunction, the spatial probability density function) of that electron? And b) maybe some experiment performed as a function of E-field would probe the degree or type of delocalization?
The field in the drift medium of an OPV is fairly large (what did we say above, ~ 10^7 v/m?). I expect that the (-) polaron’s electron “cloud” would be very polarizable in the drift (external) field. Why wouldn’t the electron density distort toward the positive electrode or plate? The distortion would be more at higher field, and vise versa. The distorted electron spatial density distribution would be at higher energy, in the absence of the applied field. The polaron would be de-stabilized. If it radiatively recombined, the emitted photon would be “redder”. But there is an external field, so will the binding energy of the field-stressed polaron be lower, the same or higher (than the zero-field binding energy)? Somewhere in here there must be something to measure….
Dr. Deibel,
thank you for this very clear review on the differences between exciton, polaron-pair and free polarons. It has a very clear/simple writing for beginner in this research field.
I have recently started working on solution processed P3HT aggregates and I may have a question regarded to polaron (any kind of them) formation: is it possible to generate polarons (pairs and/or free) that may never recombine (permanent polarons)? Without the addition of dopants or counter ions… Because I’ve seen most photoinduced absorption experiments which are suitable to observe polarons and its photophysics, but after polarons are photogenerated, they do have a characteristic lifetime which depends on the surrounding medium.
Kind regards and thanks in advance for your attention,
Renato.
Dear Renato, thanks for your message. Photogeneration creates excess carriers; they should vanish at some point in time again when the system goes back to the steady state situation before illumination / laser pulse. I have not heard of permanent polarons. Generally, however, in some semiconductor systems (inorganic, thinking of persistent photoconductivity, but potentially also organic) there are traps which can have very long release times. Regards, Carsten
Dear Dr. Deibel, thank you so much for your prompt response, it was very helpful! If you allow me, I would like to ask one other question: one consequence of polarons is the induced distortion it causes in the near environment. For polarons in conjugated polymer, is its simple existence sufficient to break the polymer conjugation at that specific position?
Thanks again!
Kind regards,
Renato.
Hmm, never thought of it that way. While I do not exactly know, I do not think so. Probably safe to think of it as distortion on top of the rest.